Get your AI Act together: The impact of the EU’s AI regulation on medical device market access
The EU AI Act will significantly impact the medical device industry, particularly in how it classifies "high-risk" devices. In this post we'll explore what it is, what it means for medical device manufacturers, and look at some of the most relevant specific requirements.
What is the EU AI Act?
The EU AI Act is a regulatory framework proposed by the European Commission to regulate Artificial Intelligence (AI) devices used or manufactured in the European Union. The goal is to ensure enhanced safety, transparency, and accountability for AI systems in the EU, and to emphasise the need for approaches that are ethical and fair. The regulation applies to anyone who makes, uses, imports, or distributes AI systems in the EU, regardless of where they are based (with certain exceptions related to military, defence, or national security purposes).
How is an AI system defined?
An AI system is a machine-based system that can operate autonomously and adapt after deployment, generating outputs like predictions or decisions based on the inputs it receives.
High-risk AI devices: a key concern for medical device manufacturers
The regulation classifies some devices as high risk. It mandates that these meet specific requirements to gain market access, and that a Notified Body review these requirements as part of a conformity assessment.
When the regulation for medical devices comes into action (currently planned for 2 August 2027), medical devices that are AI systems or that use a safety component that is an AI system, and belong to MDR risk classes II or III, will be classified as high-risk AI devices.
It is worth noting that some Class I medical devices will be classified as high risk as well, notably those with a measuring function (Class Im).
In short, all AI medical devices that require conformity assessment by a Notified Body as per EU MDR will be classified as high-risk devices.
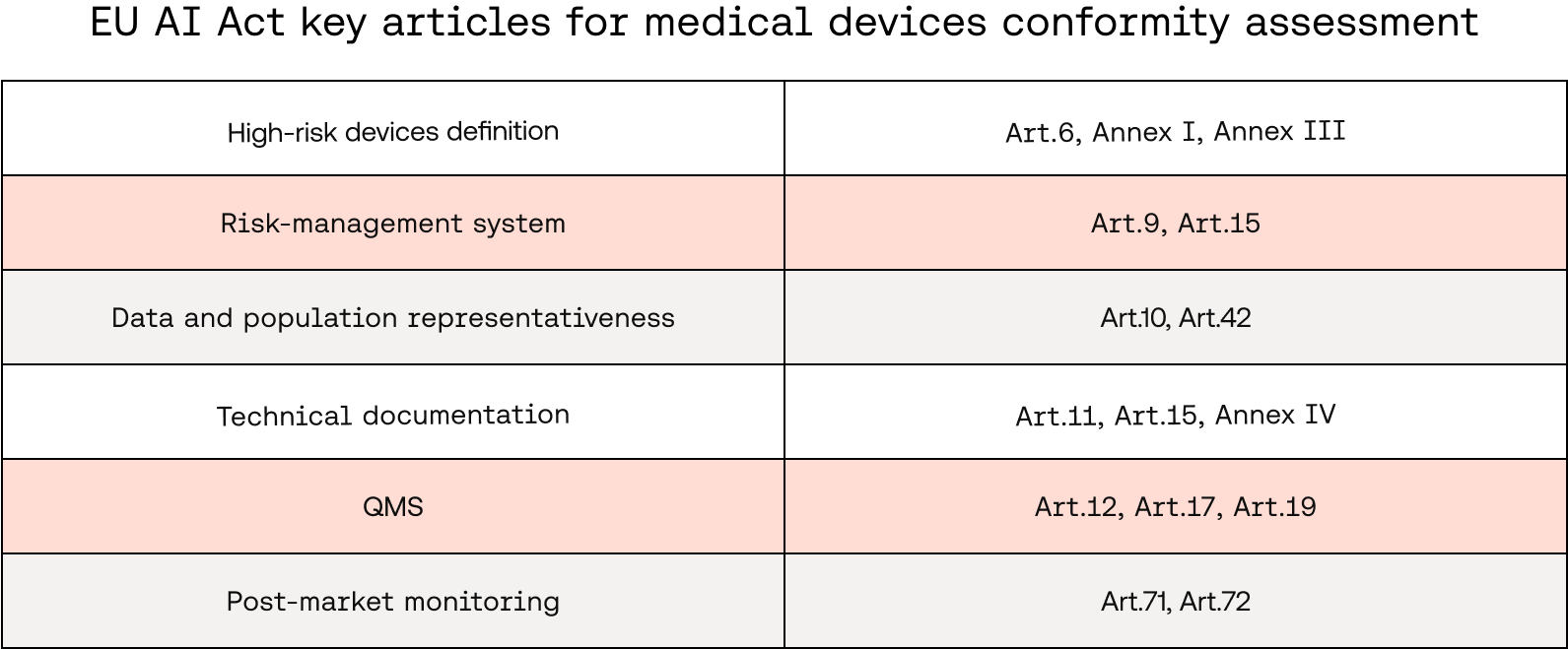
What are the obligations for high-risk AI systems?
Manufacturers are responsible for ensuring their high-risk AI systems comply with the requirements, taking into account their intended purpose as well as the generally acknowledged state of the art on AI and AI-related technologies. Those readers already familiar with MDR will recognise the formula.
There are other similarities between the requirements for high-risk devices and medical device conformity assessments. Similar to the EU MDR, the EU AI Act mandates:
- A continuous risk management system throughout the AI system life cycle
- The establishment of a Quality Management System (QMS)
- The procurement of a technical documentation file to demonstrate compliance with the requirements
Importantly, the regulation opens the door to the incorporation of an AI-specific QMS into an already existing one (required for MDR), preventing the manufacturer from having to implement two different QMS. Similarly, the AI act provides the legal basis to create and maintain a single technical documentation file containing the combined information required for both EU MDR and EU AI Act compliance, reducing the burden on manufacturers.

Specific requirements of interest for your risk management system, QMS, and technical file
Risk management system: As you would expect, this system must regularly assess risks to health and safety, and implement measures to manage these risks, ensuring any remaining risks are acceptable. Testing must be conducted to confirm the effectiveness of risk-mitigation measures.
Interestingly, the EU AI act emphasises risk management related to the fundamental rights of the people exposed to the device, which include non-discrimination and privacy rights. Manufacturers should also take measures to ensure the system is resilient against attempts by unauthorised third parties to alter its use, including through data poisoning and model poisoning.
QMS: Both medical device and AI QMS share several core elements, including the need for a regulatory compliance strategy, risk management system, design control, thorough documentation of processes and communication, and post-market monitoring. These elements ensure high standards of safety, quality, and accountability across product development and lifecycle management.
The AI Act, however, introduces a comprehensive data management QMS requirement, notably mentioning data acquisition and pre-processing, labelling, analysis, and storage. This data-centric focus reflects the acknowledgement by the EU commission of the importance of the data lifecycle within the development and marketing of an AI system.
Technical documentation: The technical documentation required for high-risk systems is comprehensive and covers several AI-specific aspects such as system and model architecture, train-validation-test procedures, performance metrics, data requirements, and cybersecurity measures.
The regulation now makes the train-validation-test approach mandatory for AI systems that incorporate the training of a machine learning model. Emphasis is placed on representativeness of the data: training, validation, and testing datasets shall be relevant, sufficiently representative, and, to the greatest extent possible, free of errors and complete with regard to the intended purpose.
While this makes complete sense for testing datasets, the requirements for training datasets to be representative of the target population can be questioned. It is common practice for the training dataset distribution to be slightly different from the target distribution. The issue may be even more obvious in cases of model pre-training, where training data can be vastly different. The judgement of Notified Bodies on these potentially grey regulatory areas will undoubtedly play an important role.
The regulation insists that the datasets used “shall take into account, to the extent required by the intended purpose, the characteristics or elements that are particular to the specific geographical, contextual, behavioural or functional setting within which the high-risk AI system is intended to be used”. In the context of medical devices, this means that the dataset used should take into account the characteristics of the intended purpose population (disease severity, clinical pathway, etc.). This requirement can be met through individual datasets or a combination thereof.
Notably the Commission will establish a simplified technical documentation form for startups and SMEs. What this form will look like and the scope of the simplification regarding the requirements remains to be seen.
A new era
The EU AI Act introduces a transformative regulatory framework that will significantly impact the medical device industry, particularly for those integrating AI into their products. While the act brings new challenges, it also provides an opportunity for manufacturers to streamline compliance by integrating AI-specific requirements into their existing QMS, risk management system, and technical documentation. Time will tell if this regulation, which is still subject to change, will strike the balance between enabling innovation and ensuring AI medical devices are safe, effective, and transparent.




Want Scarlet news in your inbox?
Sign up to receive updates from Scarlet, including our newsletter containing blog posts, sent straight to you by email.