Déjà review: leveraging your FDA work for EU MDR success
If you’ve already secured FDA clearance in the US, you're not starting from scratch when it comes to EU MDR.
Many assume that European regulatory expectations require an entirely separate development effort but in reality, there’s substantial overlap in evidence, documentation, and technical strategy. In this article, we explore key areas of convergence between FDA and EU MDR requirements, and how to make the most of the work you’ve already done, to smooth your pathway to CE marking.
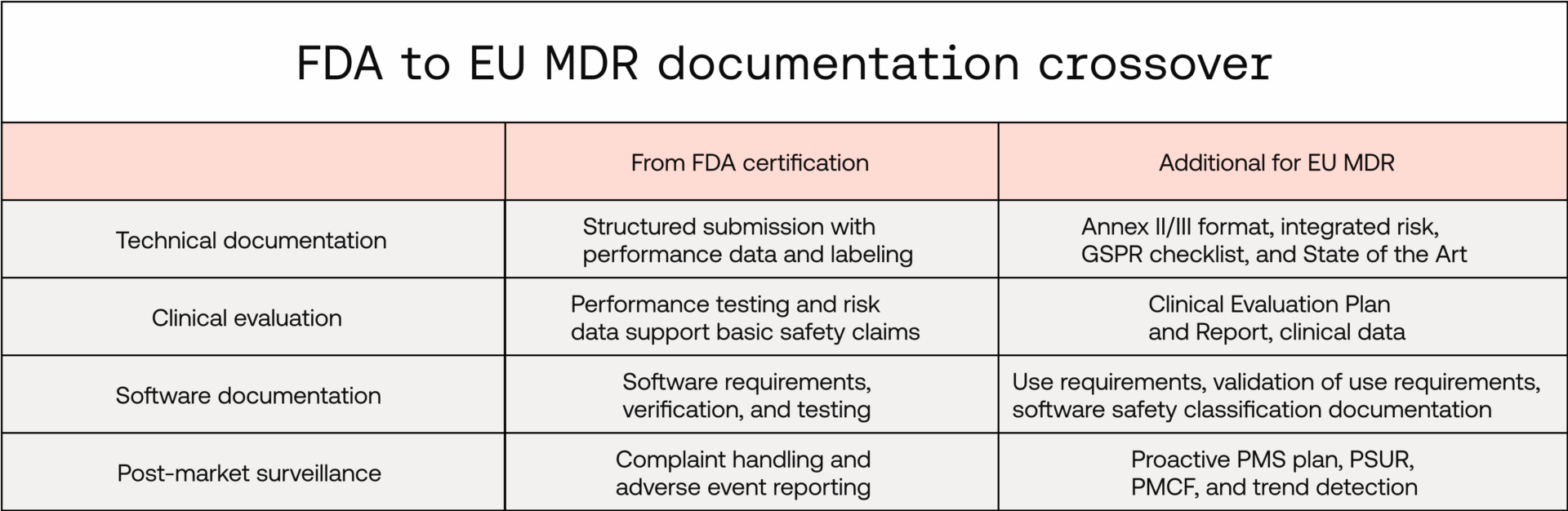
Technical documentation
The FDA’s 510(k) submission requires structured technical documentation: device description, software documentation, performance testing, and labelling. The EU MDR similarly requires a Technical Documentation File (Annex II and III), though broader in scope, and requires greater integration of risk, usability, and clinical elements. While you’ll need to add or develop a few areas, much of your documentation can be repurposed.
You can repurpose:
- Device descriptions, diagrams, and labelling
- Software architecture, specifications, and risk-control summaries
- Bench-testing and performance-testing reports
You’ll need to add or develop:
- GSPR checklist mapping safety and performance claims to evidence
- A detailed risk-management file per ISO 14971, integrated throughout
- Robust state-of-the-art determination to set evidence benchmarks
- Formal benefit-risk determination documentation
You can learn more about clinical State of the Art requirements for SaMD under EU MDR here.
Clinical evidence
The FDA’s approach relies heavily on substantial equivalence to a predicate device and performance testing while the EU MDR puts clinical benefit at the centre. Your performance testing data can go a long way to support your clinical evaluation, however, you may need to add additional clinical data to fully support your clinical benefit if this wasn’t required as part of your FDA application.
Your FDA submission likely includes data you can reuse:
- Labeling and intended-use definitions to define scope of evaluation
- Bench and performance testing to support safety and performance claims
- Human factors or usability validation to show safe use in intended environments
- Risk analysis and design controls that contribute to your clinical evaluation context
But you’ll need to add or reframe the following:
- Clinical Evaluation Plan (CEP) outlining your methodology
- Clinical data to show performance and clinical benefit
- Clinical Evaluation Report (CER) linking claims to evidence
Find out more about the types of clinical evidence required under EU MDR here.
Software development documentation
IEC 62304 and IEC 82304, which are paramount for software-development process compliance under EU MDR, are recognised consensus standards by the FDA. Many software-documentation expectations under the FDA map broadly to IEC 62304, the State-of-the-Art harmonised standard for software-development processes under EU MDR.
A key distinction is use-requirements specification and associated software validation. Use-requirements specification is not directly required by the FDA as part of 510(k) submission, and therefore not covered as part of validation.
Key areas you’ve likely already documented:
- Software requirements specification
- Software verification and testing
- Software-requirement traceability to testing
- System and software architecture design
Gaps you may need to address:
- Use-requirements specification
- Software validation of use requirements
- Explicit and granular labeling of software safety classification by item
- Greater level of documentation depth for higher software safety classification items
For more information regarding IEC 82304 software validation, check out our deep dive here.
Post-market surveillance
While the FDA focuses on complaint handling and adverse event reporting, the EU MDR takes a broader, proactive approach. Its post-market surveillance (PMS) system isn’t just a compliance exercise, it’s an opportunity to gather real-world evidence, strengthen clinical-performance claims, and continuously improve patient safety.
Your FDA PMS system is a great starting point, particularly:
- Complaint-handling workflows and internal investigations
- Adverse event investigations and reporting
Where MDR expands:
- A post-market surveillance plan tailored to your device type and risk
- Periodic Safety Update Reports (PSUR)
- A PMCF (post-market clinical follow-up) plan and report to gather real-world clinical data
- Trend reporting for non-serious incidents
- Clear documentation of Corrective Actions and Preventive Actions (CAPA)
You can read more about the particular PMCF challenges of AI medical devices here.
Equivalence claim caution
If your US submission relied on a predicate device under the 510(k) pathway, you might assume that concept carries over into Europe. But under the EU MDR, clinical equivalence has a narrower, stricter definition.
You may be able to claim equivalence under EU MDR if:
- The devices are technically and clinically similar
- The clinical condition or purpose and type of user are the same
- You can support the claim with robust scientific justification per MDCG 2020-5
You may not be able to rely on equivalence if:
- You don’t have sufficient levels of detail about the equivalent device’s technical and clinical characteristics
If your 510(k) relied heavily on a predicate device and did not include direct clinical data, you likely need to augment your clinical evaluation though a structured literature review, a clinical investigation, or both.
We've previously written about equivalence under EU MDR here.
QMS compliance and regulatory audits
If your organisation complies with the FDA’s 21 CFR Part 820 (Quality System Regulation), you already have the foundation for an EU MDR-compliant quality system. From 2 February 2026, the FDA’s Final Rule for the new Quality Management System Regulation (QMSR) will come into force, aligning with ISO 13485:2016 — as does EU MDR.
There are though a few key differences in how Quality Management Systems are assessed across the jurisdictions:
FDA inspections:
- Conducted by FDA investigators
- Regulatory compliance audit and no certification is issued
- Risk-based, typically every two years
EU MDR audits:
- Conducted by a Notified Body
- Required to pass the audit prior to CE marking
- Surveillance audits at least every 12 months, plus unannounced audits
Strategic takeaways
If you’ve gone through an FDA review, your technical and clinical groundwork is already partially laid. However, EU MDR’s structure demands more integration, more proactive lifecycle planning, and more emphasis on clinical benefit.
Instead of duplicating effort, map your FDA artifacts to MDR requirements with purpose:
- Start with your Design Dossier and reframe it using Annex II/III headings
- Extract clinical claims and link them to performance and usability data
- Integrate risk, usability, and benefit evidence into a cohesive CER
Bottom line
Beyond the regulatory milestone, your FDA clearance is also a valuable foundation for CE marking under the EU MDR. With smart planning and targeted gap analysis, your existing documentation can double up when building a compliant, comprehensive MDR submission.




Want Scarlet news in your inbox?
Sign up to receive updates from Scarlet, including our newsletter containing blog posts, sent straight to you by email.